Parsing Molecular Identifiers From the IDEaL Database, part 2
[applescript mathematica chemdraw cdx science In our last episode, we tried screen-scraping the IDEaL database to build a f-element separation database, but encountered a problem: We could not resolve 105 of molecular structure entries (i.e., we could not parse the linked ChemDraw file or the name or run a pubchem query and get a result that matched the listed molecular formula and molecular weight information). In most cases, the problem is that alkyl sidechains are only presented implicitly as molecular formulas, rather than as explicit structures so we cannot generate a proper Molecule. After initially thinking we might have to retrieve these by hand (ughh…), I had a better idea: Can the ChemDraw application correctly parse these implicit side chain specifications? It turns out that the answer is yes, and so this suggested a solution: Build the results by scripting the download, and running automated key commands in ChemDraw, to retrieve the data…
Nature of the Problem
Many of the ChemDraw files on the IDEaL database represent alkyl sidechains as abbreviated formulas rather than explicitly indicated bonds. I go into a bit more depth on this (with an explicit example) in a StackOverflow thread describing the problem. In the end, it is unclear whether one should even say this is a true bug: the CDX files are validly and completely interpreted in ChemDraw, but the CDX file does not make this information explicit enough for Mathematica, but Mathematica need not be responsible for this type of information either.
It is the case that one can use ChemDraw to export a MOL file; these are fully explicit and the resulting exported file is read correctly by Mathematica). In practice, we would have to go through each file and convert it (ugh…). Below, I will explore another strategy where we script ChemDraw.
Nature of the Solution
The ChemDraw application can interpret the correct representation, even if this is not explicitly represented in the CDX file. So the idea is to remotely control ChemDraw to get the information, and to do that we have to build three pieces of functionality:
-
Drive ChemDraw with AppleScript to open a CDX file, select all of the molecules (there should only be one…), and Edit>Copy As…>SMILES
-
Retrieve the results from the clipboard and use this to populate the entry in our record (with a sanity check against the stated molecular weight and molar mass)
-
Automate downloading the CDX files from the URLs, putting the results into a temporary directory to process them
This type of system-level application scripting is going to be very operating system dependent, so the solution below will only work on a Mac; for the record I performed this with MacOS 13.5 and ChemDraw 22.2 (and Mathematica 13.3).
Driving ChemDraw with AppleScript (via Mathematica) and getting results from clipboard
We start by creating a lightweight function wrapper around the osascript command line program which will let us run AppleScripts; this codes is copied directly from a Stack Exchange post on using AppleScript in Mathematica:
(* Code source: https://mathematica.stackexchange.com/questions/36764/how-to-import-a-numbers-spreadsheet/36772#36772 *)
AppleScript["RunFile", file_] := Run["osascript " <> file]
AppleScript["RunScript", script_] := With[
{file = ToFileName[$TemporaryDirectory, "script.txt"]},
Export[file, script, "String"];
AppleScript["RunFile", file]
]
After a bit of fumbling around, I devised the following AppleScript which takes a file path (provided as the typical POSIX filepath string) as input and results in the SMILES string on the clipboard. Once you know how to specify keystroke commands in AppleScript, it is relatively straightforward. After getting the script to run in the ScriptEditor, I created the following lightweight function wrapper using a StringTemplate:
scriptTemplate[file_] := AbsoluteFileName[file] // StringTemplate["set p to \"``\"set f to POSIX file ptell application \"ChemDraw\" open f activate tell application \"System Events\" keystroke \"a\" using {command down} keystroke \"c\" using {option down, command down} keystroke \"w\" using {command down} end tell delay 0.5end tell"]
(I found that it worked more reliably if I inserted a short delay after the copy and paste operation, as otherwise the clipboard retrieval was not always reliable.) We also need to retrieve the result from the clipboard. AFAIK, there is no built-in function for this, but some workarounds are described in this stackoverflow thread (including versions for Windows and Ubuntu; the version implemented below is for MacOS) :
(* Code source: https://mathematica.stackexchange.com/a/130224/63709 *)
Clear[getSMILESFromCDX]
getClipboard[___] := Import["!pbpaste", "Text"]
getSMILESFromCDX[file_?FileExistsQ] := getClipboard @ AppleScript["RunScript", scriptTemplate[file]]
(*demo*)
getSMILESFromCDX["~/Downloads/BuBTBP.cdx"]
(*"CCCCC(N=N1)=C(CCCC)N=C1C2=NC(C3=NC(C4=NN=C(CCCC)C(CCCC)=N4)=CC=C3)=CC=C2"*)
Automating file downloads
In our last post, we devised a function that extracts the CDX file URL from an IDEaL entry page. We will use the resulting URL to download the entry (by default, URLDownload returns a File to a temporary download directory):
(*old function*)
Clear[cdxFileURL, getMolFromEntryURL]
cdxFileURL[url_] := With[
{cdxFileURL = SelectFirst[StringContainsQ[".cdx"]] @ Import[url, "Hyperlinks"]},
URLBuild @ URLParse[cdxFileURL]
]
(*new function*)
getMolFromEntryURL[entryURL_] := With[
{downloadedFile = URLDownload @ cdxFileURL[entryURL]},
Molecule @ getSMILESFromCDX @ downloadedFile
]
(*demo*)
url = "https://www.oecd-nea.org/ideal/extractants/BuBTBP";
cdxFileURL[url]
getMolFromEntryURL[url]
(*"https://www.oecd-nea.org/ideal/structures/BuBTBP.cdx?fileKey=238"*)
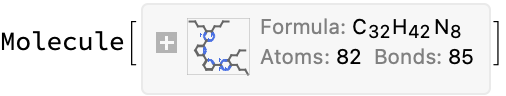
Putting it all together
Now that we have all the pieces, we just need to walk through the missing entries identified in the last post. We begin by copying/modifying some of the processing functions from the last post:
(* copied from previous post *)
massFormulaOnly[url_] := ImportString[#, "RawJSON"] &@LLMFunction["What is the molecular formula and the molecular mass in the following text? Return only the formula and number with no units or other information. Return the results as a JSON dictionary with keys molecular_formula and molecular_mass.\n``",
ProgressReporting -> False]@
StringTake[#, UpTo[1500]]& @ Import[url, "Plaintext"]
(*copied from previous post*)
moleculeAbbreviation[url_String] := Last @ Lookup["Path"] @ URLParse[url]
(*modified from previous post*)
Clear[outputData]
outputData[url_String, mol_?MoleculeQ, info_Association] := With[
{reportedMass = Interpreter["Number"]@info["molecular_mass"],
computedMass = QuantityMagnitude[mol["MolarMass"]]},
Association[
"url" -> url,
"abbreviation" -> moleculeAbbreviation[url],
"SMILES" -> mol["CanonicalSMILES"],
"InChI" -> mol["InChI"]["ExternalID"],
"InChIKey" -> mol["InChIKey"]["ExternalID"],
"computed_formula" -> mol["MolecularFormulaString"],
"reported_formula" -> info["molecular_formula"],
"computed_mass" -> computedMass,
"reported_mass" -> reportedMass,
"formula_matchQ" -> StringMatchQ[mol["MolecularFormulaString"], info["molecular_formula"]],
"mass_matchQ" -> (Round[reportedMass] == Round[computedMass])
]]
outputData[url_String, mol_, info_Association] := outputData[url, Molecule[""], info]
(*modified from previous post to use our new functions...*)
Clear[processURL]
processURL[url_String] := With[
{mol = getMolFromEntryURL[url],
info = massFormulaOnly[url]},
outputData[url, mol, info] // Append[#, ("validEntryQ" -> (#["formula_matchQ"] && #["mass_matchQ"]))] &
]
Now we apply them to the missing data (yeah, it requires making a bunch of OpenAI calls again, for an additional $0.08 USD invested in the project…).
(*read in the data*)
SetDirectory @ NotebookDirectory[];
missing = Import["2023.10.12_missing_entries.xlsx", {"Dataset", 1}, "HeaderLines" -> 1];
missingURLs = Normal @ missing[All, "url"];
newResults = processURL /@ missingURLs;
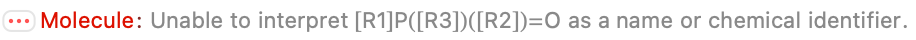
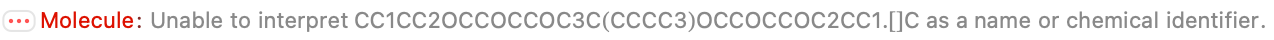
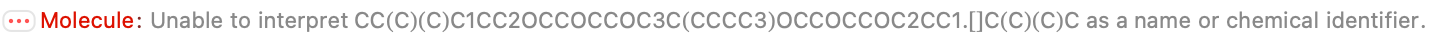



There are some parsing errors, and I guess we have to be OK with that; they look like genuine problems. But 80/105 missing entries are perfect, which shaves off a bunch of work. And most of the entries match at least one or the other of the provided formula or molecular mass, which suggests we can vet them quickly.
Length @ Select[#validEntryQ &] @ newResults
Length @ Select[#["formula_matchQ"] &] @ newResults
Length @ Select[#["mass_matchQ"] &] @ newResults
Length @ Select[#["formula_matchQ"] || #["mass_matchQ"] &] @ newResults
(*80*)
(*82*)
(*96*)
(*98*)
Conclusion
Export["2023.10.16_corrected_missing_entries.xlsx", Dataset@Select[#validEntryQ &]@newResults];
Export["2023.10.16_missing_entries_needs_proofing.xlsx", Dataset@Select[Not[#validEntryQ ] &]@newResults];
We can also merge this with the previously obtained correct results to yield a master list of valid entries:
With[
{previousResults = Import["2023.10.12_completed_ideal_molecules.xlsx", {"Dataset", 1}, "HeaderLines" -> 1],
newResults = Dataset[newResults][Select[#validEntryQ &], {"url", "abbreviation", "SMILES", "InChI", "InChIKey"}]},
Export[
"2023.10.16_all_correct_entries.xlsx",
Join[previousResults, newResults]
]];
Download these from the following links:
- 2023.10.16_corrected_missing_entries.xlsx
- 2023.10.16_missing_entries_needs_proofing.xlsx
- 2023.10.16_all_correct_entries.xlsx
Sneak preview
Tune in next time to find out how we fix these 25 errant structures. Same bat-time, same bat-channel…
ToJekyll["Parsing Molecular Identifiers From the Ideal Database, part 2", "applescript mathematica chemdraw cdx science"]